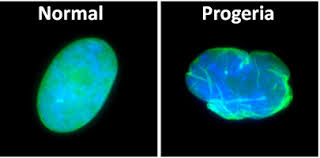
- Es una enfermedad poco común puesto que solo hay uno 103 registrados en 37 países, que recibe su nombre de su descubridor, el médico Jonathan Hutchinson en el siglo XIX,sin embargo no se ha sabido su causa hasta 2003. El síndrome Hutchinson se caracteriza por un envejecimiento acelerado de los niños.
- Casos de Progeria en el mundo:
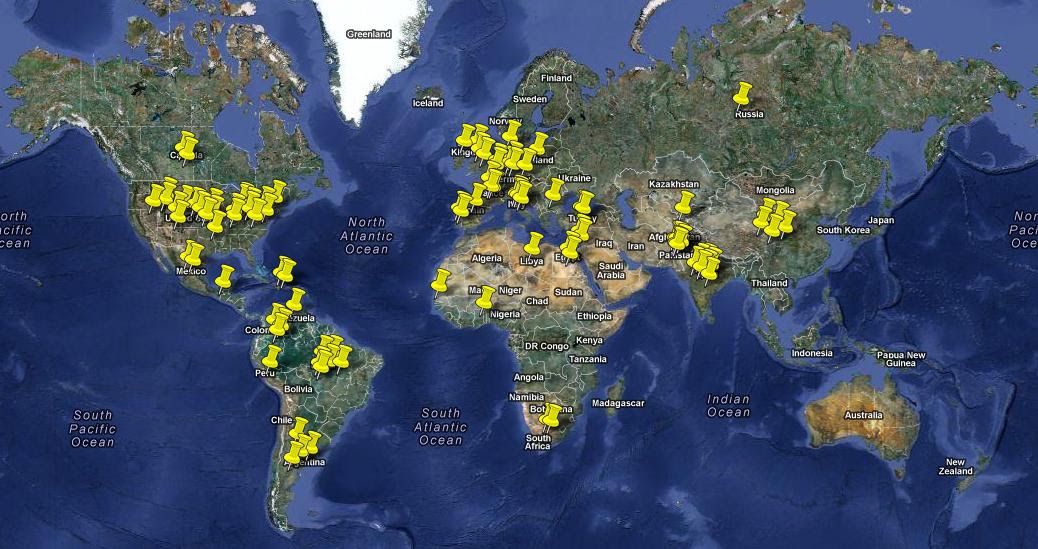
- Estadísticas de los casos de Progeria: La Progeria afecta aproximadamente a una persona por cada 4 a 8 millones de recién nacidos. Hay un estimado de 200 a 250 niños que padecen este síndrome y afecta a ambos sexos por igual y todas las razas. Se ha producido un incremento del número de casos de Progeria en el mundo:

1. Características:
Niño normal: Niño con HGPS
|
|
 |  |
2. Características principales:
- Cabeza más grande de lo normal comparada con su cara
- Nariz puntiaguda
- Piel muy fina
- Ojos grandes y prominentes
- Alopecia, incluso pueden no tener cejas o pestañas
- Dientes irregulares y amontonados
- Pecho muy estrecho
- Abdomen proyectado hacia fuera
- Las venas de la cabellera son muy prominentes y se ven fácilmente
- Piel muy pálida
- Parecen mucho más viejos de lo que son y suelen tener la piel muy seca y arrugada